publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
2024
- Conference Paper
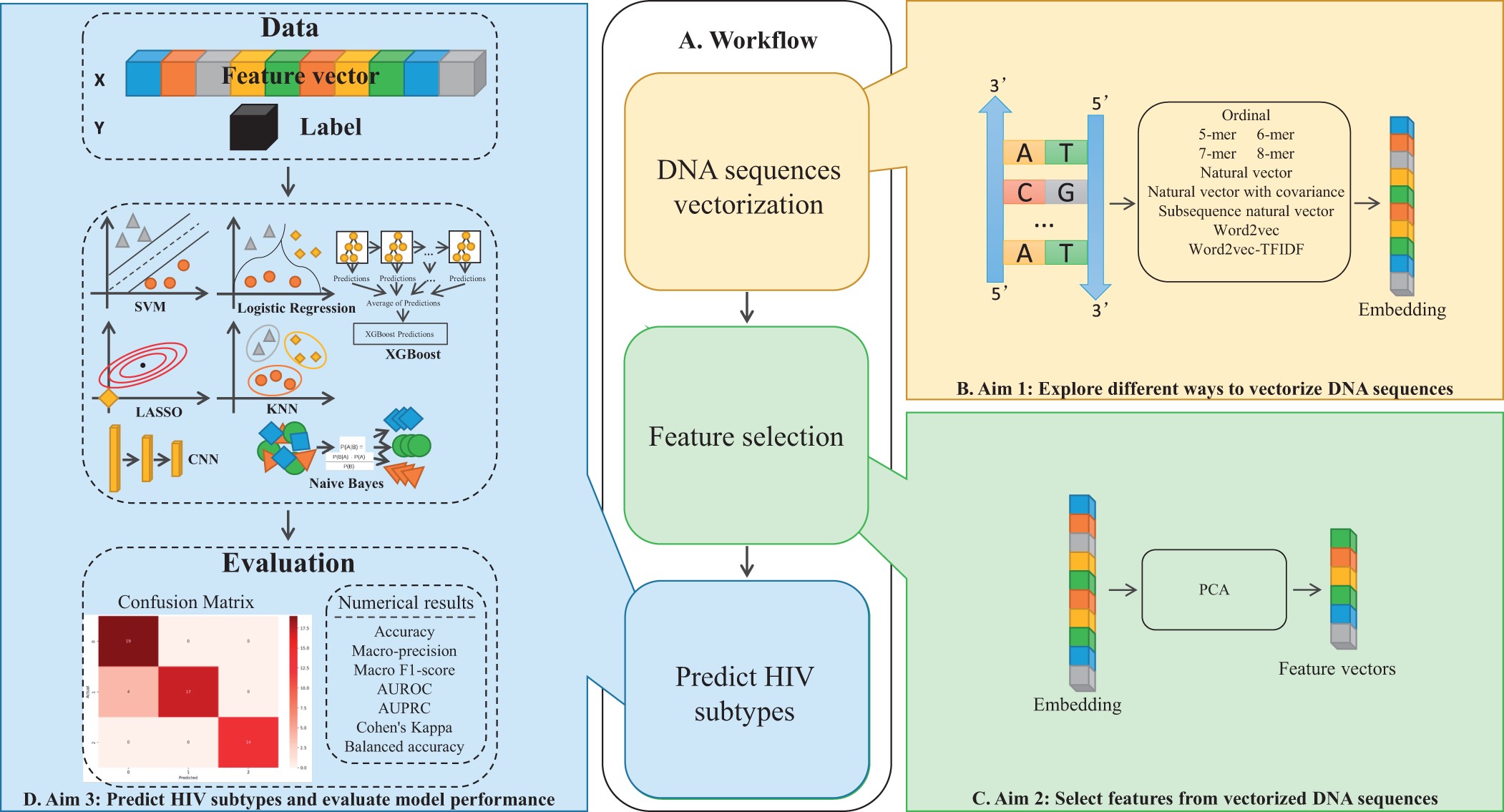 Investigating alignment-free machine learning methods for HIV-1 subtype classificationKaitlyn E Wade, Lianghong Chen, Chutong Deng, Gen Zhou, and Pingzhao HuBioinformatics Advances, 2024
Investigating alignment-free machine learning methods for HIV-1 subtype classificationKaitlyn E Wade, Lianghong Chen, Chutong Deng, Gen Zhou, and Pingzhao HuBioinformatics Advances, 2024Many viruses are organized into taxonomies of subtypes based on their genetic similarities. For human immunodeficiency virus 1 (HIV-1), subtype classification plays a crucial role in infection management. Sequence alignment-based methods for subtype classification are impractical for large datasets because they are costly and time-consuming. Alignment-free methods involve creating numerical representations or genetic sequences and applying statistical or machine learning methods. Despite their high overall accuracy, existing models perform poorly on less common subtypes. Furthermore, there is limited work investigating the impact of sequence vectorization methods, in particular natural language-inspired embedding methods, on HIV-1 subtype classification. We present a comprehensive analysis of sequence vectorization methods across machine learning methods. We report a k-mer-based XGBoost model with a balanced accuracy of 0.84, indicating that it has good overall performance for both common and uncommon HIV-1 sub-types. We also report a Word2Vec-based support vector machine that achieves promising results on precision and balanced accuracy. Our study sheds light on the effect of sequence vectorization methods on HIV-1 subtype classification and suggests that natural language-inspired encod-ing methods show promise. Our results could help to develop improved HIV-1 subtype classification methods, leading to improved individual pa-tient outcomes, and the development of subtype-specific treatments.
@article{wade2024investigating, title = {Investigating alignment-free machine learning methods for HIV-1 subtype classification}, author = {Wade, Kaitlyn E and Chen, Lianghong and Deng, Chutong and Zhou, Gen and Hu, Pingzhao}, journal = {Bioinformatics Advances}, volume = {4}, number = {1}, pages = {vbae108}, year = {2024}, publisher = {Oxford University Press}, doi = {10.1093/bioadv/vbae108}, keywords = {artificial intelligence, deep learning, HIV-1 subtyping, representation learning, classification}, url = {https://academic.oup.com/bioinformaticsadvances/article/4/1/vbae108/7723597} } - Journal Paper
 Pro portional Assist Ventilation for Mini mizing the Duration of Mechanical Ventilation (the PROMIZING study): update to the statistical analysis plan for a randomized controlled trialKaren J Bosma, Myriam Lafreniere-Roula, Arlene Jiang, Anna Heath, Yongdong Ouyang, Kaitlyn Wade, Pingzhao Hu, Karen EA Burns, Claudio M Martin, Yoanna Skrobik, and othersTrials, 2024
Pro portional Assist Ventilation for Mini mizing the Duration of Mechanical Ventilation (the PROMIZING study): update to the statistical analysis plan for a randomized controlled trialKaren J Bosma, Myriam Lafreniere-Roula, Arlene Jiang, Anna Heath, Yongdong Ouyang, Kaitlyn Wade, Pingzhao Hu, Karen EA Burns, Claudio M Martin, Yoanna Skrobik, and othersTrials, 2024We previously published the protocol and statistical analysis plan for a randomized controlled trial of Proportional Assist Ventilation for Minimizing the Duration of Mechanical Ventilation: the PROMIZING study in Trials ( https://doi.org/10.1186/s13063-023-07163-w ). This update summarizes changes made to the statistical analysis plan for the trial since the publication of the original protocol and statistical analysis plan. The Proportional Assist Ventilation for Minimizing the Duration of Mechanical Ventilation (PROMIZING) study is a multi-center, open-label, randomized controlled trial designed to determine if ventilation with proportional assist ventilation with load-adjustable gain factors will result in a shorter duration of time spent on mechanical ventilation compared to ventilation with pressure support ventilation for patients with acute respiratory failure. The statistical analysis plan for the trial was incorporated into the original publication of the protocol in Trials ( https://doi.org/10.1186/s13063-023-07163-w ) and was based on version 5.0 of the study protocol and version 1.0 of the statistical analysis plan (SAP), which included plans for both frequentist and Bayesian analyses. We have since updated the SAP to refine the Bayesian analysis plan, update the multistate model diagram, and include plans for a cluster analysis to determine if there is heterogeneity of treatment effect. This update summarizes the changes made and their rationale and provides a refined SAP for the PROMIZING trial with additional background information, in adherence with guidelines for the prospective reporting of SAPs for randomized controlled trials.
@article{bosma2024pro, title = {Pro portional Assist Ventilation for Mini mizing the Duration of Mechanical Ventilation (the PROMIZING study): update to the statistical analysis plan for a randomized controlled trial}, author = {Bosma, Karen J and Lafreniere-Roula, Myriam and Jiang, Arlene and Heath, Anna and Ouyang, Yongdong and Wade, Kaitlyn and Hu, Pingzhao and Burns, Karen EA and Martin, Claudio M and Skrobik, Yoanna and others}, journal = {Trials}, volume = {25}, number = {1}, pages = {855}, year = {2024}, publisher = {Springer}, doi = {10.1186/s13063-024-08669-7}, keywords = {bayesian; cluster; liberation; proportional assist ventilation; statistical analysis plan; ventilator-free days; weaning from mechanical ventilation.}, url = {https://link.springer.com/article/10.1186/s13063-024-08669-7} }
2023
- Journal Paper
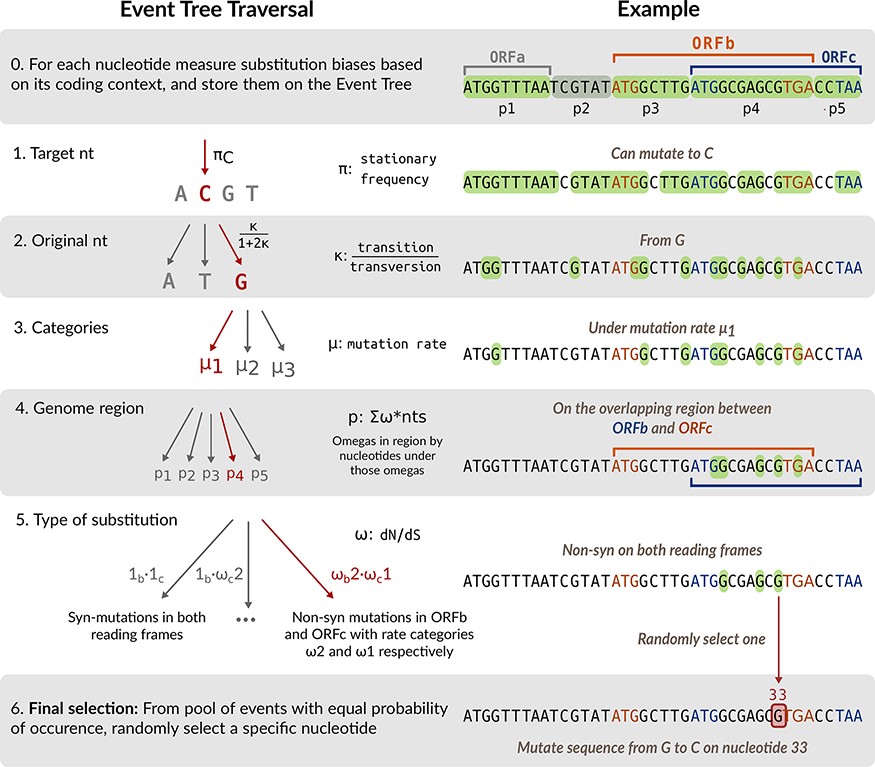 HexSE: Simulating evolution in overlapping reading framesLaura Muñoz-Baena, Kaitlyn E Wade, and Art FY PoonVirus Evolution, 2023
HexSE: Simulating evolution in overlapping reading framesLaura Muñoz-Baena, Kaitlyn E Wade, and Art FY PoonVirus Evolution, 2023Gene overlap occurs when two or more genes are encoded by the same nucleotides. This phenomenon is found in all taxonomic domains, but is particularly common in viruses, where it may provide a mechanism to increase the information content of compact genomes. The presence of overlapping reading frames (OvRFs) can skew estimates of selection based on the rates of non-synonymous and synonymous substitutions, since a substitution that is synonymous in one reading frame may be non-synonymous in another and vice versa. To understand the impact of OvRFs on molecular evolution, we implemented a versatile simulation model of nucleotide sequence evolution along a phylogeny with any distribution of open reading frames in linear or circular genomes. We use a custom data structure to track the substitution rates at every nucleotide site, which is determined by the stationary nucleotide frequencies, transition bias and the distribution of selection biases (dN/dS) in the respective reading frames. Our simulation model is implemented in the Python scripting language. All source code is released under the GNU General Public License version 3 and are available at https://github.com/PoonLab/HexSE.
@article{munoz2023hexse, title = {HexSE: Simulating evolution in overlapping reading frames}, author = {Mu{\~n}oz-Baena, Laura and Wade, Kaitlyn E and Poon, Art FY}, journal = {Virus Evolution}, volume = {9}, number = {1}, pages = {vead009}, year = {2023}, publisher = {Oxford University Press UK}, doi = {10.1093/ve/vead009}, url = {https://academic.oup.com/ve/article/9/1/vead009/7023538} }
2022
- Journal Paper
 Revisiting the recombinant history of HIV-1 group M with dynamic network community detectionAbayomi S Olabode, Garway T Ng, Kaitlyn E Wade, Mikhail Salnikov, Heather E Grant, David W Dick, and Art FY PoonProceedings of the National Academy of Sciences, 2022
Revisiting the recombinant history of HIV-1 group M with dynamic network community detectionAbayomi S Olabode, Garway T Ng, Kaitlyn E Wade, Mikhail Salnikov, Heather E Grant, David W Dick, and Art FY PoonProceedings of the National Academy of Sciences, 2022The prevailing abundance of full-length HIV type 1 (HIV-1) genome sequences providesan opportunity to revisit the standard model of HIV-1 group M (HIV-1/M) diversitythat clusters genomes into largely nonrecombinant subtypes, which is not consistentwith recent evidence of deep recombinant histories for simian immunodeficiency virus(SIV) and other HIV-1 groups. Here we develop an unsupervised nonparametricclustering approach, which does not rely on predefined nonrecombinant genomes, byadapting a community detection method developed for dynamic social network analysis.We show that this method (dynamic stochastic block model [DSBM]) attains a signif-icantly lower mean error rate in detecting recombinant breakpoints in simulated data(quasibinomial generalized linear model (GLM), P < 8 × 10−8), compared to otherreference-free recombination detection programs (genetic algorithm for recombinationdetection [GARD], recombination detection program 4 [RDP4], and RDP5). Whenthis method was applied to a representative sample of n = 525 actual HIV-1 genomes,we determined k = 29 as the optimal number of DSBM clusters and used change-pointdetection to estimate that at least 95% of these genomes are recombinant. Further,we identified both known and undocumented recombination hotspots in the HIV-1 genome and evidence of intersubtype recombination in HIV-1 subtype referencegenomes. We propose that clusters generated by DSBM can provide an informativeframework for HIV-1 classification.
@article{olabode2022revisiting, title = {Revisiting the recombinant history of HIV-1 group M with dynamic network community detection}, author = {Olabode, Abayomi S and Ng, Garway T and Wade, Kaitlyn E and Salnikov, Mikhail and Grant, Heather E and Dick, David W and Poon, Art FY}, journal = {Proceedings of the National Academy of Sciences}, volume = {119}, number = {19}, pages = {e2108815119}, year = {2022}, publisher = {National Academy of Sciences}, doi = {10.1073/pnas.2108815119}, keywords = {HIV-1, recombination, virus evolution, stochastic block model, dynamic network}, url = {https://www.pnas.org/doi/10.1073/pnas.2108815119} }
2021
- Journal Paper
 CoVizu: Rapid analysis and visualization of the global diversity of SARS-CoV-2 genomesRoux-Cil Ferreira, Emmanuel Wong, Gopi Gugan, Kaitlyn Wade, Molly Liu, Laura Muñoz Baena, Connor Chato, Bonnie Lu, Abayomi S Olabode, and Art FY PoonVirus Evolution, 2021
CoVizu: Rapid analysis and visualization of the global diversity of SARS-CoV-2 genomesRoux-Cil Ferreira, Emmanuel Wong, Gopi Gugan, Kaitlyn Wade, Molly Liu, Laura Muñoz Baena, Connor Chato, Bonnie Lu, Abayomi S Olabode, and Art FY PoonVirus Evolution, 2021Phylogenetics has played a pivotal role in the genomic epidemiology of severe acute respiratory syndrome coronavirus 2, such as tracking the emergence and global spread of variants and scientific communication. However, the rapid accumulation of genomic data from around the world—with over two million genomes currently available in the Global Initiative on Sharing All Influenza Data database—is testing the limits of standard phylogenetic methods. Here, we describe a new approach to rapidly analyze and visualize large numbers of SARS-CoV-2 genomes. Using Python, genomes are filtered for problematic sites, incomplete coverage, and excessive divergence from a strict molecular clock. All differences from the reference genome, including indels, are extracted using minimap2 and compactly stored as a set of features for each genome. For each Pango lineage (https://cov-lineages.org), we collapse genomes with identical features into ‘variants’, generate 100 bootstrap samples of the feature set union to generate weights, and compute the symmetric differences between the weighted feature sets for every pair of variants. The resulting distance matrices are used to generate neighbor-joining trees in RapidNJ that are converted into a majority-rule consensus tree for each lineage. Branches with support values below 50 per cent or mean lengths below 0.5 differences are collapsed, and tip labels on affected branches are mapped to internal nodes as directly sampled ancestral variants. Currently, we process about 2 million genomes in approximately 9 h on 52 cores. The resulting trees are visualized using the JavaScript framework D3.js as ‘beadplots’, in which variants are represented by horizontal line segments, annotated with beads representing samples by collection date. Variants are linked by vertical edges to represent branches in the consensus tree. These visualizations are published at https://filogeneti.ca/CoVizu. All source code was released under an MIT license at https://github.com/PoonLab/covizu.
@article{ferreira2021covizu, title = {CoVizu: Rapid analysis and visualization of the global diversity of SARS-CoV-2 genomes}, author = {Ferreira, Roux-Cil and Wong, Emmanuel and Gugan, Gopi and Wade, Kaitlyn and Liu, Molly and Baena, Laura Mu{\~n}oz and Chato, Connor and Lu, Bonnie and Olabode, Abayomi S and Poon, Art FY}, journal = {Virus Evolution}, volume = {7}, number = {2}, pages = {veab092}, year = {2021}, publisher = {Oxford University Press UK}, doi = {10.1093/ve/veab092}, keywords = {SARS-CoV-2, genomic epidemiology, data visualization}, url = {https://academic.oup.com/ve/article/7/2/veab092/6431771} }